AI-ML Drug Discovery Platform
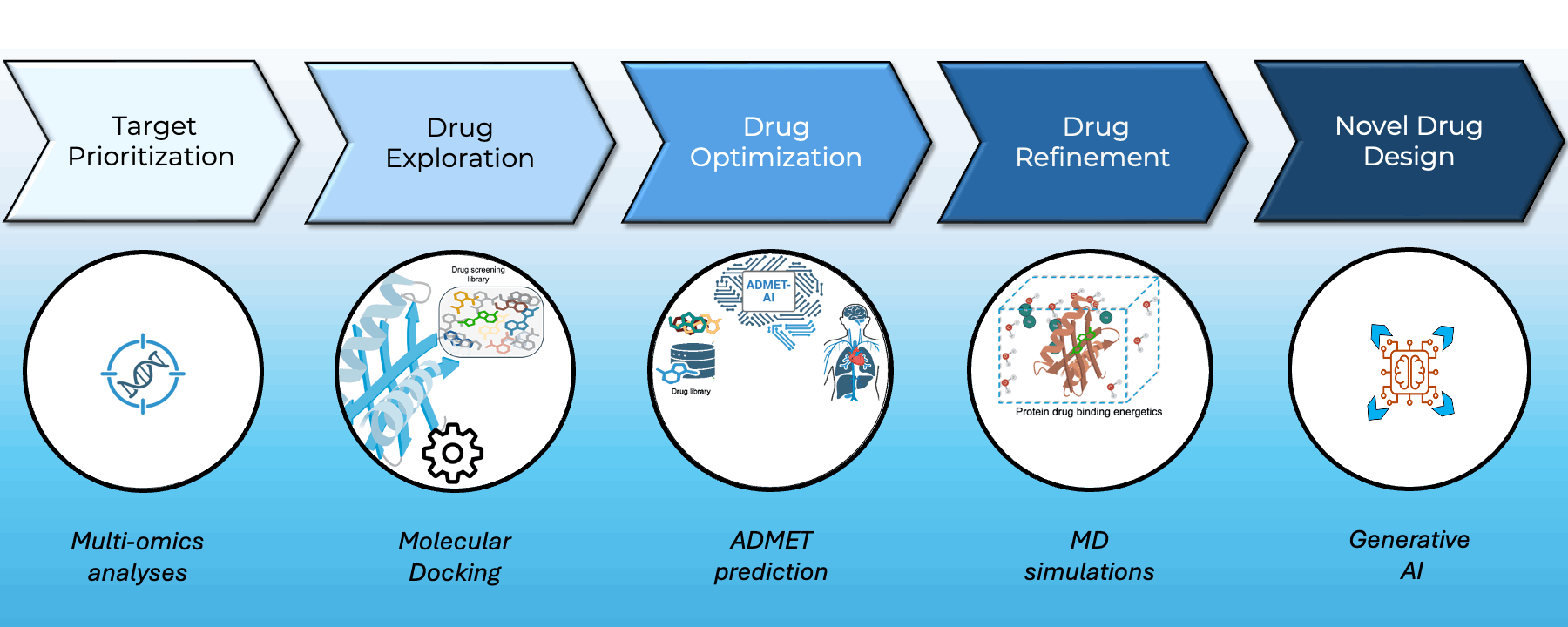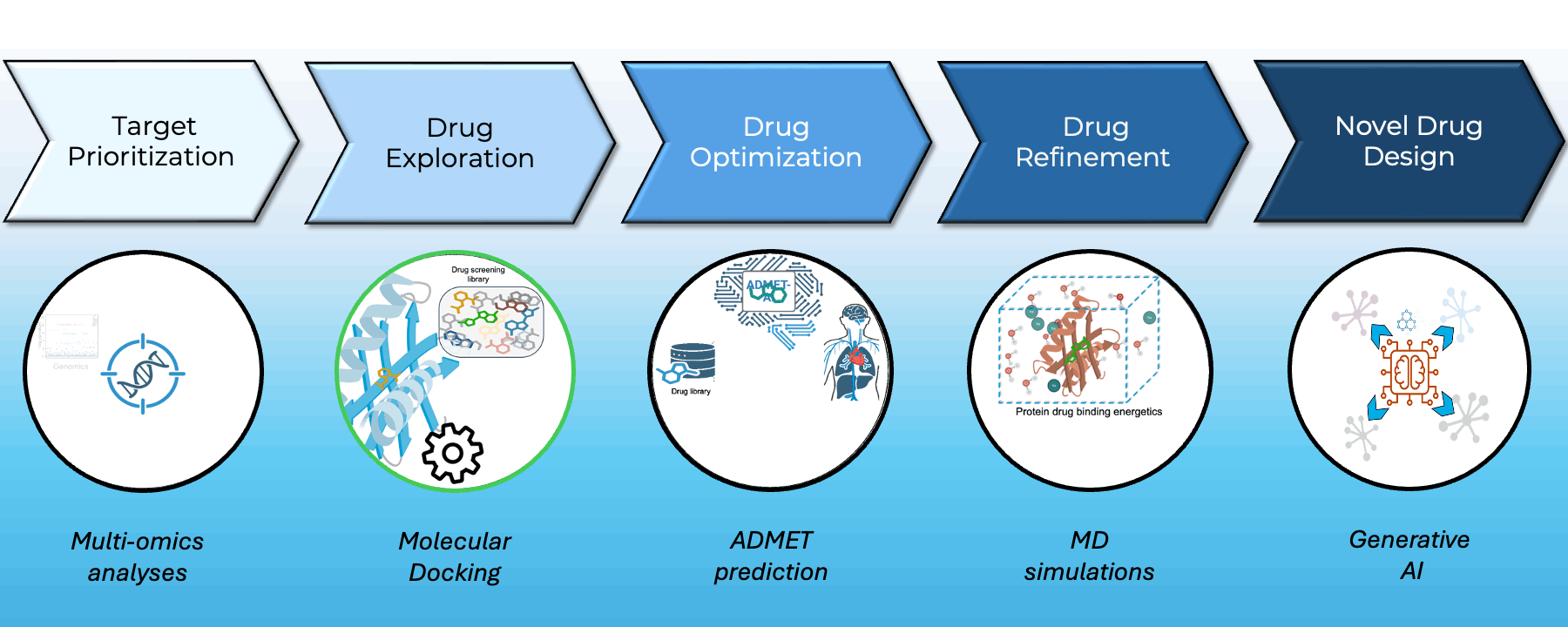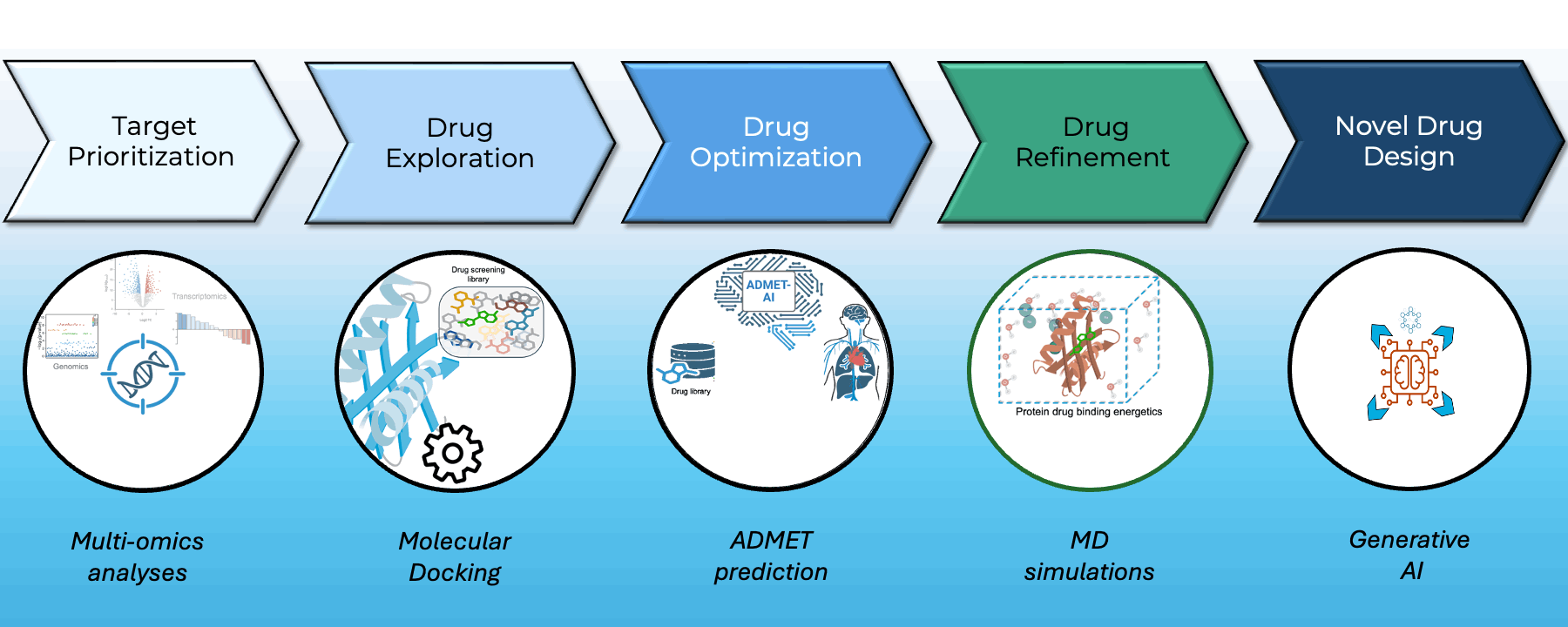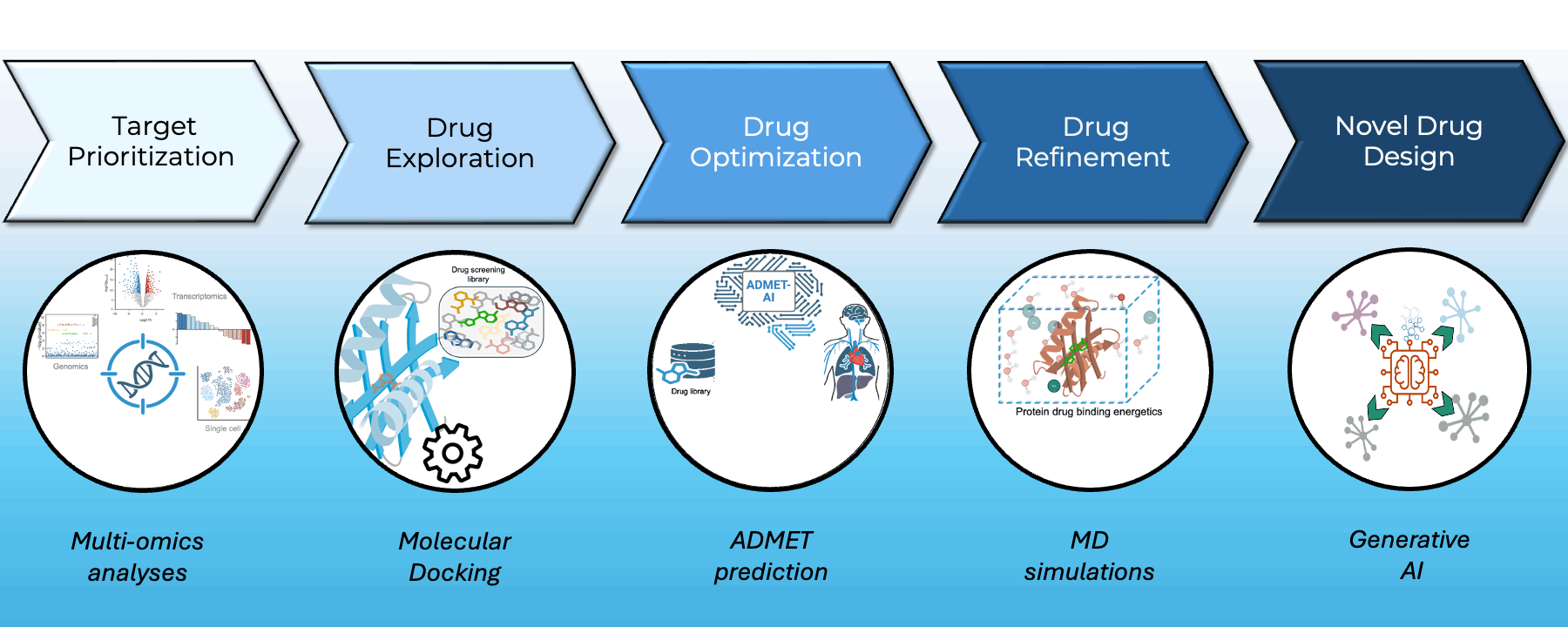
At Greenstone Biosciences, we have built an end-to-end AI-driven drug discovery pipeline that integrates our proprietary iPSC-based multi-omics data with state-of-the-art computational modeling.
Key Capabilities:
- Target Prioritization: AI models trained on multi-omics datasets from our iPSC biobank identify novel therapeutic targets.
- Virtual Screening: High-throughput docking using AutoDock Vina and BOLTZ2 across major libraries such as ApexBio and ZINC to find optimal small-molecule binders.
- Drug Property Prediction: In-house ADMET-AI platform (Swanson et al., Bioinformatics) delivers one of the fastest, most accurate ADMET predictors — available at admet.ai.greenstonebio.com.
- Cardiotoxicity Modeling: ADMET-AI DICT (Mukherjee et al., Circulation, 2025) predicts drug-induced cardiotoxicity using FDA-labeled datasets (> 1,000 drugs).
- Organ-Specific Models: Custom predictors for liver, lung, and hematologic toxicity extend safety profiling.
- Molecular Dynamics: Candidate refinement via OpenMM and Gromacs simulations ensures high-affinity, stable target binding before synthesis.
Why it matters:
Our platform unites AI, biobank data, and molecular simulation to accelerate discovery, reduce attrition, and prioritize safer, more effective therapies.
Databases
- PubMed — biomedical literature database
https://pubmed.ncbi.nlm.nih.gov/
- UniProt — protein sequence & function database
https://www.uniprot.org/
- RCSB PDB — Protein Data Bank (3D structures of proteins & nucleic acids)
https://www.rcsb.org/
- KEGG — Kyoto Encyclopedia of Genes and Genomes (pathways, drugs, genomes)
https://www.genome.jp/kegg/
- STRING — protein–protein interaction networks
https://string-db.org/
Reactome — curated biological pathways
https://reactome.org/
Chemical / Bioactivity Databases
- ChEMBL — curated bioactivity dataset (ligand–target binding, assays) https://www.ebi.ac.uk/chembl/
- BindingDB — protein–ligand binding affinities https://www.bindingdb.org/
- PubChem — compound repository https://pubchem.ncbi.nlm.nih.gov/
- ZINC — virtual screening database http://zinc.docking.org/
- Therapeutics Data Commons (TDC) — ML-ready datasets https://tdcommons.ai/
Molecular Dynamics Simulations
- GROMACS — high-performance biomolecular MD http://www.gromacs.org/
- OpenMM — GPU-accelerated MD with Python APIs http://openmm.org/
Docking Tools
- AutoDock Vina — molecular docking engine https://github.com/ccsb-scripps/AutoDock-Vina
- BOLTZ-2 — open-source generative AI model for drug design (from MIT & collaborators)
https://boltz.ai/ (GitHub: https://github.com/jwohlwend/boltz)
RNA-seq and Multi-Omics Tools
- STAR — RNA-seq aligner https://github.com/alexdobin/STAR
- DESeq2 — differential expression (R) https://bioconductor.org/packages/release/bioc/html/DESeq2.html
- edgeR — differential expression (R) https://bioconductor.org/packages/release/bioc/html/edgeR.html
- limma-voom — RNA-seq linear modeling (R) https://bioconductor.org/packages/release/bioc/html/limma.html
- Seurat — scRNA-seq analysis (R) https://satijalab.org/seurat/
- Scanpy — scRNA-seq analysis (Python) https://scanpy.readthedocs.io/
ADMET and Toxicity Prediction
- SwissADME — physicochemical & ADMET properties http://www.swissadme.ch/
- ADMET-AI — large-scale AI ADMET prediction https://admet.ai.greenstonebio.com
General Cheminformatics and AI Libraries
- RDKit — cheminformatics library https://www.rdkit.org/
- DeepChem — ML library for chemistry/biology https://github.com/deepchem/deepchem
- OpenFold — open-source protein structure prediction https://github.com/aqlaboratory/openfold
RFdiffusion — generative protein modeling https://github.com/RosettaCommons/RFdiffusion
